ISO 13485 en pratique : construire un Système Qualité Dispositifs Médicaux opérationnel (avec modèle Excel)
ISO 13485 est la norme internationale de référence pour les systèmes de management de la qualité des dispositifs médicaux. Au-delà d’un simple manuel et de procédures écrites, elle exige une gestion fondée sur des preuves, une maîtrise des risques selon la norme ISO 14971, des contrôles de la conception et de la production, ainsi qu’une amélioration continue.
Cet article propose une mise en œuvre concrète, qui s’appuie sur un modèle Excel prêt à l’emploi pour structurer les enregistrements et maintenir le cap. Bien que le chemin soit parfois escarpé, un suivi rigoureux de ces directives permettra d’atteindre de nouveaux sommets en termes de qualité et de sécurité des dispositifs médicaux.
1) Les piliers d’ISO 13485 (vision synthétique)
- Leadership & gouvernance : politique qualité, responsabilités, ressources, culture documentaire.
- Conception & développement (Design Control) : DHF, revues, V&V, transfert en prod, DMR.
- Gestion des risques (ISO 14971) : danger → situation dangereuse → dommage, évaluation S×P, contrôles, résiduel & acceptabilité.
- Production & maîtrise des procédés : qualification IQ/OQ/PQ, validation, maintenance & étalonnages, environnement.
- Traçabilité & DHR : lots/SN, composants, libération, enregistrements de fabrication.
- Achats & fournisseurs : qualification, criticité, audits, revues périodiques.
- NC & CAPA : corrections, causes racines, actions correctives, vérification d’efficacité.
- Réclamations & vigilance : évaluation, déclaration réglementaire, lien CAPA.
- Compétence & formation : exigences, enregistrements, efficacité.
- Maîtrise documentaire : versions, approbations, revues périodiques.
- Audits internes & Revue de direction : performance du QMS, décisions, ressources.
- Mesure & amélioration : objectifs, KPI, tendances, preuves factuelles.
2) Mettre en musique la norme avec le modèle Excel
Contexte & Périmètre
Cadrez le périmètre QMS, identifiez parties intéressées (autorités, ON, patients, distributeurs), fixez vos exclusions justifiées et rôles (Direction, Qualité, PRRC).
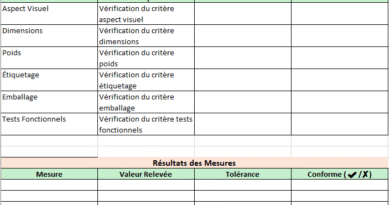
Design Control : DHF → DMR → DHR
- DHF (Conception_DHF) : entrées/sorties de conception, revues, V&V, transfert.
- DMR (DMR) : référence « maître » des spécifs produit/procédés/contrôles/IFU.
- DHR (Traçabilité_DHR) : dossier de lot/numéro de série, composants, équipements, libération.
Gestion des risques (ISO 14971)
- Risques_Produit(14971) : saisissez Sévérité (1–5) et Probabilité (1–5) → score inhérent calculé ; documentez contrôles et résiduel ; statuez Acceptabilité (Acceptable / Non acceptable / Conditionnelle).
- Bon réflexe : relier chaque risque à une sortie de conception ou à une exigence de contrôle.
Production & procédés spéciaux
- Validation_Procédé : IQ/OQ/PQ, revalidation due automatisée (rappel).
- Équipements_Étalonnage : prochaine échéance & retard signalé.
Chaîne d’approvisionnement
- Fournisseurs : criticité, due diligence, audits ; Revue_due si > 12 mois.
Non-conformités, CAPA, réclamations & vigilance
- NC_CAPA : type, gravité, 5-Pourquoi/ISHIKAWA, action corrective, efficacité vérifiée ; retard auto.
- Réclamations_Vigilance : gravité, évaluation médicale, déclaration réglementaire requise (Oui/Non), lien CAPA ; âge > 15 j mis en évidence.
Compétences & documents
- Formations_Compétence : exigences/réalisations, en retard si échéance dépassée.
- Documents : versions, approbations, revue périodique (alerte automatique).
Pilotage
- Objectifs_Qualité : indicateurs, cibles, statut.
- Tableau_de_Bord : CAPA en retard, réclamations > 15 j, audits/changements/étalonnages/formations/documents en retard, fournisseurs à revoir, total risques.
3) Démarche de déploiement (90 jours)
J0–J30 — Structurer
- Politique qualité, rôles, périmètre QMS.
- Arborescence documentaire et modèles (procédure, enregistrement).
- Lancer l’inventaire risques (14971) pour les produits prioritaires.
- Ouvrir DHF des projets actifs ; constituer DMR minimum viable.
J31–J60 — Exécuter
- Mettre à niveau les procédures clés : maîtrise des docs/enreg., NC/CAPA, réclamations, achats/fournisseurs, validation, contrôle en cours, traçabilité, formation.
- Déployer IQ/OQ/PQ pour les procédés spéciaux.
- Activer Fournisseurs (éval initiale + plan d’audit).
- Renseigner DHR sur les lots pilotes.
J61–J90 — Prouver & améliorer
- Audit interne : constats & CAPA.
- Revue de Direction : objectifs, KPI, ressources, décisions.
- Boucler DMR, tracer DHF → DMR → DHR.
- Stabiliser Tableau_de_Bord et routines de suivi.
4) Indicateurs qui comptent (exemples)
- Taux de NC/million d’unités, % CAPA en retard, délai moyen clôture CAPA.
- Conformité étalonnages (% à l’heure), procédés validés vs prévus.
- Réclamations par 1 000 unités, taux de MDR/FDA reportable, délai de réponse.
- Achats : % fournisseurs critiques revus/audités.
- Formation : % compétences conformes au poste.
- Design : couverture V&V des exigences, écarts résiduels, dérives.
5) Préparer l’évaluation de l’Organisme Notifié
- Dossier prêt-audit :
- DHF/DMR/DHR reliés et complets.
- 14971 : matrice risques, contrôles, justification de l’acceptabilité.
- Validation procédés & étalonnages (preuves réelles).
- CAPA : preuves de vérification d’efficacité.
- Réclamations/Vigilance : traçabilité jusqu’aux actions.
- Audits internes + Revue de Direction récentes et factuelles.
- Échantillons : un lot complet, un changement significatif, un CAPA majeur, un fournisseur critique, un enregistrement de formation.
6) Pièges fréquents (et comment les éviter)
- Risque « cosmétique » : scores sans lien avec des décisions → ancrez chaque risque à un contrôle ou à une exigence.
- CAPA non efficaces : absence d’évaluation d’efficacité → définissez un critère mesurable à l’ouverture.
- Validation procédés incomplète : IQ/OQ sans PQ sur matière/équipement représentatifs.
- Traçabilité lacunaire : composants non liés au lot/SN → verrouillez le DHR.
- Maîtrise documentaire statique : revues périodiques non tenues → utilisez les alertes d’échéance.
7) Comment utiliser efficacement le modèle
- Paramétrez les “Listes” (statuts, gravité, acceptabilité, etc.).
- Conception : alimentez DHF, créez le DMR, démarrez 14971.
- Production : complétez DHR, Validation_Procédé, Équipements_Étalonnage.
- Support : faites vivre NC_CAPA, Réclamations_Vigilance, Fournisseurs, Formations, Documents.
- Pilotez via Tableau_de_Bord et animez un rituel hebdo (30 min) : CAPA/retards/risques critiques.
ISO 13485 — Modèle Excel QMS ergonomique (v2) : clair, moderne et prêt à l’audit
Utile: un QMS selon ISO 13485 est pragmatique: c’est la preuve quotidienne qui lie la conception, les risques, la production, et l’amélioration. Le modèle excel vous aide à structurer vos preuves, automatiser vos rappels, visualiser les priorités et accélérer la préparation audit. La conformité n’est plus une charge, mais un flux qui sauvegardent les patients, les autorités, votre entreprise.
Ce classeur vous aide à piloter un QMS conforme ISO 13485 (dispositifs médicaux) de la conception à la production, avec des listes déroulantes, des calculs automatiques, des alertes de retard et un tableau de bord KPI.
2) Comment il est organisé (onglets clés)
Listes
Menus déroulants normalisés (Statut, Oui/Non, Priorité, Gravité, Type NC/Changement, Formation, Acceptabilité, etc.).
➡️ Astuce : adaptez ces listes à votre vocabulaire.
Contexte_&_Périmètre
Périmètre QMS, parties intéressées, exigences réglementaires, rôles/responsabilités.
➡️ But : poser le cadre d’audit.
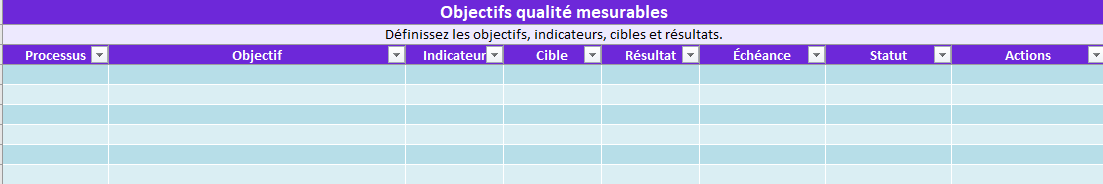
Objectifs_Qualité
Objectifs, indicateurs, cibles, résultats, échéances, statut, actions.
➡️ Suivi : ce tableau alimente vos revues de direction.
Risques_Produit(14971)
Danger → situation dangereuse → dommage, Sévérité × Probabilité = Risque (inhérent/résiduel), Acceptabilité.
➡️ Automatisation : calculs de risque, mise en couleur.
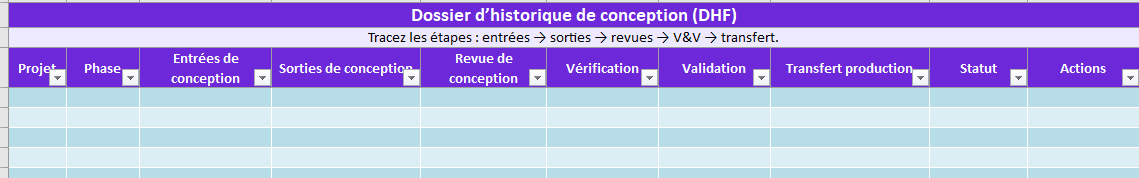
Conception_DHF
Entrées/sorties de conception, revues, V&V, transfert en production.
➡️ Traçabilité : reliez aux risques 14971.
DMR / Traçabilité_DHR
- DMR : références maîtres (spécifs, procédés, contrôles, IFU, emballage).
- DHR : lot/SN, composants, équipements, libération.
➡️ Preuves : dossiers prêts pour l’audit.
Changements
Type, justification, impact risque, validation (IQ/OQ/PQ si besoin), retard (jours) automatique.
➡️ Contrôle : évitez les dérives non validées.
NC_CAPA
NC → correction → cause racine → action corrective → efficacité vérifiée ; retard calculé.
➡️ Efficacité : pensez critère de succès à l’ouverture.
Réclamations_Vigilance
Gravité, évaluation médicale, déclaration réglementaire requise (Oui/Non), lien CAPA, âge (jours).
➡️ Signal : > 15 jours mis en évidence.
Audits
Interne/externe, critères, constats/NC, actions de suivi, retard.
➡️ Rythme : programmez à l’année.
Fournisseurs
Criticité, évaluations/audits, Revue_due si > 12 mois.
➡️ Chaîne d’appro : focalisez sur les critiques.
Formations_Compétence
Exigences par rôle, réalisations, en retard si échéance dépassée.
➡️ Conformité : montrez la compétence au poste.
Documents
Versions, approbations, prochaine revue et alerte de retard.
➡️ Maîtrise documentaire simplifiée.
Tableau_de_Bord
KPI prêts : CAPA en retard, réclamations > 15 j, audits/changements/étalonnages/formations/documents en retard, fournisseurs à revoir, total risques.
➡️ Pilotage : une page pour décider.
3) Ce qui a été amélioré (style & UX)
- Police : Calibri lisible (titres 16 pt, sous-titres 11 pt, en-têtes 11 pt gras, corps 10 pt).
- Lisibilité : texte à retour à la ligne automatique, alignements soignés.
- Ergonomie : gel des volets sur A4 pour garder les en-têtes visibles quand on défile.
- Thème : esthétique violette conservée (contraste renforcé sur les en-têtes).
Mode d’emploi express (3 minutes)
- Paramétrez “Listes” (vocabulaire maison).
- Renseignez DHF/DMR/DHR pour vos produits et Risques_Produit(14971).
- Faites vivre NC_CAPA, Réclamations, Changements, Audits, Fournisseurs, Formations, Documents.
- Pilotez via Tableau_de_Bord (traitez d’abord les retards et risques élevés).

ISO 13485 — Feuille de route claire vers la certification (en 16 étapes)
Phase 1 — Préparer (construire le QMS)
- Définir le périmètre & le contexte
Produits/process concernés, sites, exclusions justifiées, parties intéressées (autorités, ON, clients), exigences applicables. - Gouvernance & plan de projet
Sponsor Direction, Responsable Qualité/PRRC, RACI, jalons, budget, politique qualité. - Cartographier les processus
Processus “cœur” (conception, achats, production, contrôle, vigilance) + support (doc, compétences, maintenance, IT); interactions et indicateurs. - Maîtrise documentaire
Procédures minimales (contrôle des docs/enregistrements, NC/CAPA, réclamations, achats/fournisseurs, conception, traçabilité/DHR, validation procédés, étalonnages, formation). - Gestion des risques (ISO 14971) intégrée
Méthode S×P, critères d’acceptabilité, liens vers exigences de conception/production; registre risques produit. - Conception & développement (Design control)
Démarrer/mettre à niveau DHF (entrées/sorties, revues, V&V, transfert) et DMR (références maîtres). - Infrastructure & environnement
Zones propres si pertinent, équipements qualifiés, maintenance, métrologie (étalonnages planifiés).
Phase 2 — Mettre en œuvre (faire fonctionner)
- Achats & fournisseurs
Criticité, évaluation initiale, contrats & exigences, audits/ revues périodiques. - Production & validation des procédés
IQ/OQ/PQ pour procédés spéciaux (stérilisation, nettoyage, soudure contrôlée…), maîtrise du changement. - Traçabilité & DHR
Identification produit, lots/SN, composants, enregistrements de fabrication, libération. - Contrôle produit & surveillance
Contrôles réception / en cours / final, maîtrise du produit non conforme, révisions d’accès/limites. - Compétences & sensibilisation
Matrice compétences par poste, formation initiale & recyclage, efficacité attestée. - NC & CAPA, réclamations & vigilance
Ouverture → cause racine → actions → vérification d’efficacité; gestion réclamations et déclarations réglementaires si nécessaire. - Mesure & audits internes
KPI (NC, CAPA, plaintes, délais, étalonnages, validation), programme d’audits internes couvrant tout le périmètre. - Revue de direction
Entrées: résultats d’audits, KPI, réclamations/NC, performances fournisseurs, ressources, risques.
Sorties: décisions, priorités, moyens.
Phase 3 — Faire certifier (audits tiers)
- Certification tierce partie
- Choisir l’organisme (accrédité) et caler le planning.
- Stage 1 (documentaire) : périmètre, conformité des procédures, préparation Stage 2.
- Corrections ciblées des écarts Stage 1.
- Stage 2 (sur site) : efficacité réelle des processus & enregistrements (échantillons DHR, CAPA, réclamations, validations…).
- Clôture des NC (preuves d’efficacité), décision puis certificat (cycle 3 ans).
- Surveillance annuelle (Années 1 & 2), recertification en Année 3.
Dossier “prêt-audit” (checklist express)
- Politique qualité, périmètre, cartographie processus.
- DHF/DMR/DHR reliés et complets.
- 14971 : matrice risques, contrôles, justification de l’acceptabilité.
- Validation procédés (IQ/OQ/PQ), étalonnages à jour.
- Achats/fournisseurs : criticité, évaluations, audits, actions.
- NC & CAPA : preuves de vérification d’efficacité.
- Réclamations/Vigilance : traçabilité→CAPA.
- Audits internes & Revue de direction récents, KPI suivis.
Chrono indicatif (peut varier selon taille/complexité)
- Semaines 0–4 : périmètre, gouvernance, docs clés, cartographie, démarrage 14971 & DHF/DMR.
- Semaines 5–10 : déploiement procédures, validation procédés, fournisseurs, DHR pilotes.
- Semaines 11–16 : KPI en routine, audit interne, revue de direction.
- Semaines 17–20 : Stage 1 + corrections.
- Semaines 21–26 : Stage 2, clôture NC, décision de certification.